How Rfam families are built
SEED alignments and secondary structure annotation
Rfam Seed alignment (SEED) contains a curated subset of representative sequences for each family, derived from the scientific literature, expert databases, or specialist knowledge of non-coding RNAs. The SEED alignment contains a secondary structure annotation, which represents the conserved secondary structure for these sequences.
The ideal basis for a new family is an RNA element that:
has some known functional classification
is evolutionarily conserved
has evidence for a secondary structure
To build a new family, we must first obtain at least one experimentally validated example from the published literature. If any other homologues are identified in the literature, we will add these to the SEED. Alternatively, if these are not available, we will try to identify other members either by similarity searching (using Infernal) or manual curation.
Where possible, we will use a multiple sequence alignment and secondary structure annotation provided in the literature. If this is the case, we will cite the source of both the alignment and the secondary structure. You should note that the structure annotations obtained from the literature may be experimentally validated or they may be RNA folding predictions (commonly MFOLD). Unfortunately, we do not discriminate between these two cases when we cite the PubMed Identifier (PMID), and you will need to refer to the original publications to clarify.
Alternatively, where this information is not available from the literature, we will generate an alignment and secondary structure prediction using various software, such as RNAfold or RNAalifold. This software allows us to cherry-pick the best alignment and secondary structure prediction. Historically, the methods used to make these alignments and folding predictions have varied. Author names added to the list indicate that the published or predicted secondary structure has been manually curated in some way. The last author on the list will be the most recent editor of the secondary structure. You can find the method we have used for the SEED alignment or the secondary structure annotation in the SE and SS lines of the Stockholm format or in the curation information pages.
From the SEED alignment, we use the Infernal software to build a Covariance model (CM) for this family. This model is then used to search the Rfamseq database for other possible homologs.
Expanding the SEED (iteration)
If the CM search of rfamseq identifies any homologs that we believe would improve the SEED, we use the Infernal software (cmalign) to add these sequences to the SEED alignment. From the new SEED, the CM is re-built and re-searched against rfamseq. We refer to this process of expanding the SEED using Infernal searching as iteration. We continue to iterate the SEED until we have good resolution between real and false hits and cannot improve the SEED membership further.
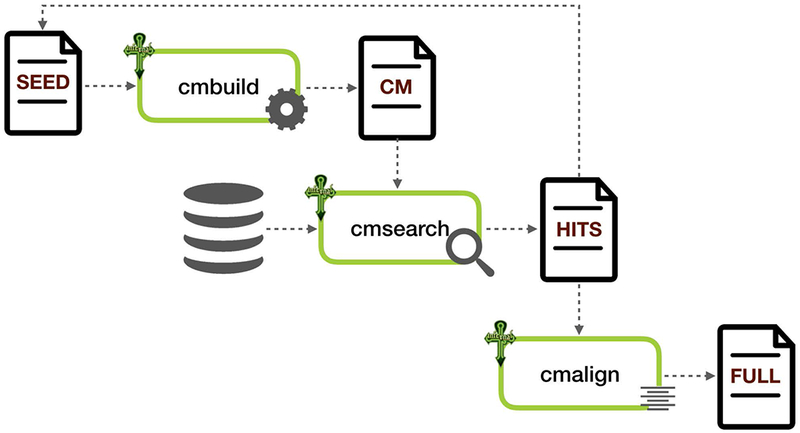
Building an RNA family using Infernal
Important points to remember about SEED alignments
We can only build families using the sequences in rfamseq.
We can only build a family where we can identify more than one sequence in rfamseq.
Sequences in the SEED cannot be manually altered in any way, e.g. no manual excision of introns, no editing of sequencing errors, no marking up modified nucleotides, etc.
At least one sequence in the SEED will have some experimental evidence of transcription, e.g. northern blot or RT-PCR, and preferably, some evidence of function.
The secondary structure should ideally have experimental support (such as structure probing, NMR, or crystallography) and/or evidence of evolutionary conservation. However, when biochemical or structural evidence is not available, computationally predicted secondary structures are also generated and used.
Rfam FULL alignments
The Rfam Full alignment (FULL) contains all of the sequences in Rfamseq that we can identify as members of the family. The alignment is generated by searching the covariance model for the family against the Rfamseq database. Matches that score above a Gathering cutoff are aligned to the CM to produce the FULL alignment. All sequences in the SEED will also be present in the FULL alignment.
Wikipedia annotations
To provide some background and functional information about a family, we link to a Wikipedia page that provides relevant background information on the family. We have either linked to an existing page or created the page ourselves in Wikipedia. As this annotation is hosted by Wikipedia, you are free to edit, correct, and otherwise improve this annotation, and we would encourage you to do so.
Phylogenetic trees
All our phylogenetic trees are generated using fasttree.