Frequently Asked Questions
Documentation
What are “SEED” and “FULL” alignments?
There are two kinds of Rfam multiple sequence alignments:
1. The SEED alignment is a hand-curated alignment of known members of the family. This alignment may not contain all known members of a family, but rather a representative set. We use the Infernal software to build a covariance model from this alignment.
The covariance model is used to search the Rfamseq sequence database for other family members and build a FULL alignment including all instances of a family.
What do the scores for hits to Rfam models mean?
When you search a sequence against Rfam and obtain a hit to one of our families, we report the start and end coordinates of the matching region, the orientation of the match, and the bit score. The bit score (also known as the log-odds score ) is generated by the Infernal software when it tries to match your sequence to the model. In very simple terms, it is a measure of how well your sequence matches the model; the higher the bit score, the better your sequences fits the model.
More specifically the bit score is the log2 of the probability of the query sequence given the model, over the probability of the sequence given the null model:
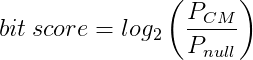
In theory this means that positive bits scores are significant but, in practice, more conservative cutoffs are used as the size of the database means we can observe hits with low positive bits scores by chance. (See the Infernal user guide for more information.)
Where does your secondary structure annotation come from?
Ideally, when we build a SEED alignment, the initial secondary structure annotation is obtained from the literature. In these cases the secondary structure is usually available only for a few of the member sequences in the SEED. Our aim is to generate models that represent conserved secondary structure, so when we begin to expand the membership of the SEED to be as representative as possible, we will only retain the secondary structure annotation that is conserved between the majority of sequences. You should also note that the annotations obtained from the literature may be experimentally validated or they may be RNA folding predictions. We do not discriminate between the two and you will need to refer to the original publications to clarify.
In those cases where no secondary structure prediction is available in the literature, but where we have good set of SEED sequences, we use RNAalifold from The ViennaRNA Package for aligning the sequences following a secondary structure as a reference.
You can find the alignment and structure source for each family in the curation tab, or in the SE and SS lines in the Stockholm file. Where the source is obtained from the literature, we will provide the PubMed identifier (PMID). You should also note that the SEED alignments often get updated between releases and may be manually adjusted by the curator. As a result, attempts to obtain the same structure using the same prediction method, may not return exactly the same structure as shown on the Rfam SEED alignment. We usually indicate where the a structure has been manually edited.
What is your definition of an RNA family?
We will group sequences into a “family” where we can identify sequence or secondary structure conservation using our covariation models. This is decided when we build our SEED alignment and search the CM against rfamseq. From the resulting searches we decide where the cutoff threshold should be.
When we set this cut off threshold, we are essentially deciding that any sequences that score above the threshold are true, homologous members of the family, whilst those below are “chance hits”. This discrimination between true and false is usually very clear if we have a representative SEED alignment.
Occasionally, for various biological reasons, it can be extremely difficult to get good resolution between true and false predictions. In such case we make an informed decision on where the cutoff should be. As a result, some families may contain false positives (often pseudogenes) or may also lose some true positives below the threshold. In such cases we will have made the best choice we can in order to limit the false positive and loss of true positives. If you have queries about the membership of any of our families, please Contact us and we will try to clarify or resolve the problem.
How can I tell which are predicted and which are experimentally confirmed sequences?
Unfortunately, it is not currently possible to do this, since we do not add a source tag to each individual sequence in either our SEED or FULL alignments. All of our families (SEED alignments) are based on one or more experimentally validated exemplars of the family, but the majority of the other member sequences are added by homology search and manual curation. We have high confidence in these members of the SEED alignment that we use to build the covariance model and computationally predict other possible members in the nucleotide database.
You can study the descriptions of sequences extracted from the EMBL nucleotide database, occasionally this contains useful information about function.
Why is my favourite sequence not in the family?
The most likely reason is that it is not in the EMBL release that rfamseq is based on. With each major release, e.g. 14.0, 15.0, we update the underlying nucleotide database. You can check which version we are currently using here. If, however, your sequence is in the relevant EMBL release but is still absent from a relevant family, it is possible that our model may need to be improved. Please Contact us with the relevant information and we will decide whether the sequence should indeed be included and, if so, we will try to improve our model.
Where can I find out more about RNA sequence analysis/covariance models/SCFGs?
The Infernal software package, which is an essential companion to the Rfam database, now has extensive documentation, along with some description of how covariance models work for RNA sequence analysis. Background and theory can also be found in the excellent book Biological Sequence Analysis by Richard Durbin, Sean Eddy, Anders Krogh, and Graeme Mitchison (Cambridge University Press, 1998). For more references see Citing Rfam.
Searching
How can I find information about a particular RNA family?
You can do this in several ways. If you already know the Rfam accession or name of the family, you can use the “jump to” boxes on the home page or any tabbed page in the website. Alternatively, if you’re not sure of the family accession or correct name and want to try a broad-ranging search, you should use the “keyword” search box in the header of each page. This search allows the use of ambiguous terms and will search multiple sections of the database for a match to your query term. The results page will give you a list of all the families with matches and you can follow the links to the summary page for each family.
If you’re not even sure of your query term and simply want to browse our families, click on the “browse” link in the header of every page. This takes you to an index that lists all Rfam families according to accession and ID and links directly to the summary page for each family.
How can I search my DNA sequence for non-coding RNA genes?
Both our single sequence and batch searches allow you to search a nucleotide sequences against the Rfam model library. Any hits to Rfam families will be returned with start and end coordinates, orientation and a score for each hit.
For short single sequences, our single sequence search tool will return Rfam matches to your sequence interactively. However, if your sequence is longer than 2Kbp, we suggest that you fragment it into smaller, overlapping segments and use the batch search facility. You might find this tool useful for splitting large sequences into fragments.
Finally, if you have a very large number of sequences to search, you may find it most convenient to download and run Rfam locally (see section Genome annotation for more information).
Downloading
What do the sequence identifiers in your alignments mean?
The identifier “AY033236.1/563-353” means that the EMBL accession is “AY033236”, the sequence version is “1” (optional), the start coordinate is “563” and the end coordinate is “353”, the strand is given by the order of the coordinates, in this case it is negative.
How can I view or download a family alignment?
From the family summary page, go to the “Alignments” tab. The alignments tab will give you multiple drop down options on how to either view or download the SEED sequences for this family, in an aligned or fasta format. The formatting options allow you to select which type of format you would prefer.
If the alignment is very large the formatting tool may not be suitable and you may prefer to use the preformatted alignment in Stockholm format. A number of Stockholm alignment re-formatters and viewers exist, such as the sreformat program from the HMMer package and the RALEE major mode for Emacs. You can read more about Stockholm format on Wikipedia.
If you are interested retrieving alignments for multiple families, you can download all our SEED alignments in Stockholm format flat-files, and the covariance models used to generate them, from our ftp site.
How can I download a domain-specific subset of covariance models?
The rfam-taxonomy project on GitHub contains a list of domain-specific Rfam families. For example, you can access Bacteria-specific or Virus-specific Rfam families and generate a domain-specific subset of covariance models that can be used with the Infernal software to scan genomes or any other sequences.
How can I download a subset of sequences from a family?
Unfortunately, this has not been implemented yet. There are plans in place to modify the underlying Rfam database to allow this.
How can I download all Rfam sequences for my favourite species?
Unfortunately, this has not been implemented yet. Please Contact us if you need help.
The “Taxonomy” tab on the search page will allow you to perform taxonomic queries. This function also allows you to search with queries from internal nodes of the NCBI taxonomic tree. However, the results are only returned on the family level, not the sequence level.
Rfam and Infernal
How do I filter Infernal output by Rfam family type?
Sometimes it is useful to filter Infernal output based on Rfam family type, for example, if you are only interested in rRNA families.
1. Get a list of Rfam families for each RNA type (see Search by entry type).
For example, selecting the rRNA checkbox gives the following list:
RF00001 5S_rRNA Gene; rRNA RF00002 5_8S_rRNA Gene; rRNA RF00177 SSU_rRNA_bacteria Gene; rRNA RF01118 PK-G12rRNA Gene; rRNA RF01959 SSU_rRNA_archaea Gene; rRNA RF01960 SSU_rRNA_eukarya Gene; rRNA RF02540 LSU_rRNA_archaea Gene; rRNA RF02541 LSU_rRNA_bacteria Gene; rRNA RF02542 SSU_rRNA_microsporidia Gene; rRNA RF02543 LSU_rRNA_eukarya Gene; rRNA RF02545 SSU_trypano_mito Gene; rRNA RF02546 LSU_trypano_mito Gene; rRNA RF02547 mtPerm-5S Gene; rRNA RF02554 ppoRNA Gene; rRNA RF02555 hveRNA Gene; rRNA
Create a file on your computer called
rfam-ids.txtwith a list of Rfam ids:
RF00001 RF00002 RF00177 RF01118 RF01959 RF01960 RF02540 RF02541 RF02542 RF02543 RF02545 RF02546 RF02547 RF02554 RF02555Tip
If you would like to download the list of RNA families and types as text, click Show the unformatted list at the bottom of the search results page. Then copy and paste into an editor and save the file for example as
rfam-types.txt. You can then create therfam-ids.txtfile with the commandcat rfam-types.txt | awk '{ print $1 }' > rfam-ids.txt.
Use the grep command to filter Infernal results.
For instance, given an Infernal tblout file
results.tblout(example file), run this command:grep -f rfam-ids.txt results.tbloutIt will print only the lines from
results.tbloutthat contain Rfam ids specified inrfam-ids.txt.Alternatively, if you want to exclude some families from your analysis, you can use the following command:
grep -v -f rfam-ids.txt results.tbloutThis will print only the lines that do not contain Rfam ids listed in
rfam-ids.txt.
You can use this procedure to filter Infernal results by any set of Rfam families. For example, you can get a list of Rfam families using Taxonomy search and get Infernal search results from families found in a specific taxonomic group.
Other
I would like to submit a family
Great! We are very keen for the community to help keep us updated on new families. Ideally, a new family for Rfam should contain elements (RNA sequences) that have some known functional classification, are evolutionarily conserved and have evidence for a secondary structure. The families should not solely be based on prediction only, e.g. RNAz, EvoFold, or QRNA predictions, nor solely on transcriptomic data, e.g. tiling array or deep sequencing. For more detailed information on how to submit a family, please read the rest of the Rfam documentation but, if you have any queries, please do Contact us.
If your family is sufficiently interesting, or if you have several of them, you may be interested in publishing your family in the RNA families track that is available through the RNA Biology journal.
How can I edit a SEED alignment?
We do not currently provide public access to edit our alignments. This is advantageous in that it maintains our standard of alignments and structures, but, if you feel our SEED alignment/structure annotations can and should be improved, please Contact us, preferably supplying us with a new alignment, in Stockholm format, and we will do our best to incorporate the improvements.