Glossary
Clan
An Rfam clan is a group of families that either share a common ancestor but are too divergent to be reasonably aligned or a group of families that could be aligned, but have distinct functions. For example, the LSU clan (CL00112) includes 5 families describing different types of large ribosomal subunit RNAs, including bacterial, eukaryotic, and archaeal LSU families.
Browse Rfam clans or download a list of families belonging to clans (Rfam.clanin) that can be used by Infernal for automatic clan competition.
Clan competition
When several families from the same clan match the same sequence region (for example, both Sarbecovirus 5’ UTR and the bCoV-5UTR families match the 5’ UTR sequence of SARS-CoV-2), the redundancy can be removed based on the bit scores and lengths of the matching sequences. The process of redundancy removal is called clan competition.
All sequences on the Rfam website have already undergone this process, but if you perform searches using Infernal, you may want to skip any potentially redundant hits (see the --oskip option of the cmalign program in the Infernal manual).
ClustalW
A general purpose multiple sequence alignment program for DNA (RNA) which we use while building our SEED alignments. See the Clustal web server.
Covariance model (CM)
A secondary structure profile for a RNA structural alignment (also called profile stochastic context-free grammars). The Rfam covariance models are used by the Infernal software to find instances of RNA families in RNA or DNA sequences. Find more about Covariance models and stochastic context-free grammars.
DESC file
Each family is described using in a DESC file that includes the information such as family description, database references, RNA type, and publications (see the tRNA DESC file as an example).
Family
A group of RNA sequences which are believed to be evolutionarily related in sequence or secondary structure.
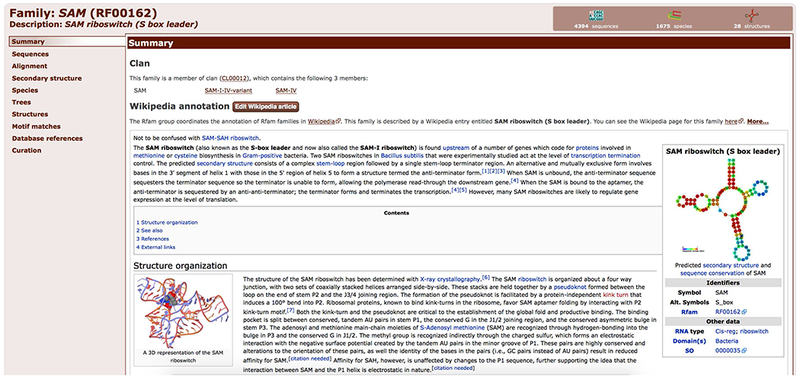
SAM riboswitch Rfam family
Full alignment (FULL)
An alignment of the set of related sequences that score higher than the manually set threshold values for the covariance model of a particular Rfam family.
For details about generating a full alignment, see the Rfam Curr Protoc Bioinformatics paper.
Gathering cutoff
The bit score gathering threshold (GA cutoff), set by Rfam curators when building the family. All sequences that score at or above this threshold will be included in the full alignment and are believed to be true homologs to the model. For more information, see Nawrocki et al., 2015.
Infernal
Infernal is the core software that enables us to make consensus RNA secondary structure profiles (covariance models (CMs)) for our families. We also use Infernal for searching sequence databases for homologous RNAs. See the Infernal website for more details.
MFOLD
RNA structure prediction algorithm which utilises minimum free energy information. See the MFOLD publication.
Pfold
RNA folding software which folds alignments using a Stochastic Context-Free Grammars (SCFG) trained on rRNA alignments. It takes an alignment of RNA sequences as input and predicts a common structure for all sequences. See the Pfold publication.
Rfamseq
Rfamseq Rfamseq is a representative, reduced-redundancy set of genome sequences against which ncRNA family models are searched to evaluate phylogenetic distribution and define gathering thresholds. It provides the sequence space used to assess ncRNA family models across phylogenetic diversity. Starting with Rfam 13.0, Rfamseq is based on a collection of complete, non-redundant, and representative genomes maintained by UniProt <http://www.uniprot.org/proteomes>. The more recent reported Rfamseq version is reported in Rfam 15.0. See the Rfam 15.0 publication.
rfamseq is usually updated with each major Rfam release, e.g., 14.0 or 15.0. You can find out the information about rfamseq currently in use in the README file in the Rfam FTP archive.

The taxonomic distribution of cellular organisms in Rfamseq 15 organized by the phylogenetic kingdom.
RNAalifold
Folds pre-computed alignments using a combination of free-energy and covariation measures. Part of the Vienna package.
RNAfold
Predicts minimum free energy structures and base pair probabilities from single RNA sequences. Part of the Vienna package.
R-scape
R-scape is a method for testing whether covariation analysis supports the presence of a conserved RNA secondary structure in a multiple sequence alignment. R-scape is used to create and improve Rfam families, and R-scape visualisations are shown on the secondary structure tab for each family (for example, SAM riboswitch).
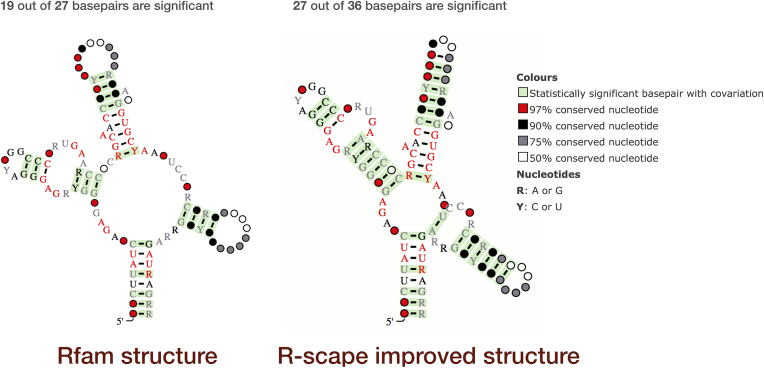
R-scape visualisation of SAM riboswitch
Seed alignment (SEED)
A manually curated sample of representative sequences for a family. These sequences are aligned and annotated with a consensus secondary structure. This alignment is used to build the covariance model for the family. See SEED alignments and secondary structure annotation for more information.

An example of SEED alignment view of Rfam family RF00164 implemented with the Nightingale framework.
Sequence region
A single segment of nucleotide sequence in our alignments. Multiple sequence regions from a single EMBL sequence may be in the same family.
Stockholm format
A multiple sequence alignment format used by Rfam (and Pfam) for the dissemination of protein and RNA sequence alignments. For more information see the Wikipedia article on Stockholm format or the Rfam tRNA alignment.
Type
A representative functional classification used to organise Rfam families into RNA types. This ontology does not currently directly relate to the ontologies used by other databases. For a full list of RNA types see the Search by entry type section.
WUSS format
The Washington University Secondary Structure (WUSS) format is designed to make it easier to see the secondary structure by eye and follows the following conventions:
Symbol |
Meaning |
|
basepairs in simple stem loops |
|
basepairs enclosing multifurcations |
|
internal loops and bulges |
|
single strand between helices |
|
single stranded residues external to any secondary structure |
|
insertions relative to the consensus |